Finding rare (un-annotated) cell types with DRVI#
Rare cell types / states are often missing from the annotations of a dataset: there are too few of them to form their own cluster, so they get absorbed into a neighboring cell type’s label. DRVI, however, tends to give such a population its own latent dimension, because the population has a distinct gene program.
In this notebook we define a rare-cell-type dimension by three criteria and use them to scan the DRVI latent space of the immune dataset:
Rare activity — Only a small fraction of cells are active on the dimension.
Spatial cohesion — The active cells sit close together in the latent manifold, rather than being scattered.
Interpretability — The dimension has a significant gene program signature.
The immune dataset (human PBMC / bone marrow) has no Fibroblast annotation. Yet a handful of
fibroblast contaminant cells are present. We will discover them as a dimension that meets the three
criteria, use the interpretability scores to hypothesize their identity, and finally prove it with
the canonical fibroblast marker COL1A1 — a gene that was not among the 2000 highly variable
genes (HVGs) the model was trained on, but is present in the full data.
We reuse the already-trained model from the general DRVI pipeline — there is no retraining here.
Contact#
For questions and help requests, you can reach out in the scverse discourse.
If you found a bug, please use the issue tracker.
Install#
This notebook uses the tutorials extra of drvi-py (DRVI plus helper packages such as
leidenalg, used here for Leiden clustering). Install it once in your environment with:
pip install "drvi-py[tutorials]"
On Colab, the next cell does this for you. Remove it if your environment is already set up.
import sys
import subprocess
# if branch is stable, will install via pypi, else will install from source
branch = "latest"
IN_COLAB = "google.colab" in sys.modules
if IN_COLAB and branch == "stable":
subprocess.check_call([sys.executable, "-m", "pip", "install", "drvi-py[tutorials]"])
elif IN_COLAB and branch != "stable":
subprocess.check_call([sys.executable, "-m", "pip", "install",
"git+https://github.com/theislab/drvi.git#egg=drvi-py[tutorials]"])
Imports#
import warnings
warnings.filterwarnings("ignore")
from pathlib import Path
import numpy as np
import pandas as pd
import anndata as ad
import scanpy as sc
from scipy.spatial.distance import pdist
import scvi
import drvi
from drvi.model import DRVI
print("Last run with scvi-tools version:", scvi.__version__)
print("Last run with DRVI version:", drvi.__version__)
Last run with scvi-tools version: 1.4.3
Last run with DRVI version: 0.2.6
# Making plots prettier
sc.set_figure_params(dpi=100, frameon=False, figsize=(3, 3))
from matplotlib import pyplot as plt
plt.rcParams["figure.dpi"] = 100
plt.rcParams["figure.figsize"] = (3, 3)
Config#
We load the artifacts produced by the general training/interpretability pipeline:
drvi_model— the trained DRVI model.embed.h5ad— the latent space (one column per latent dimension) with dimension stats inembed.varand pre-computed interpretability scores inembed.varm.Immune_HVG_human.h5ad— the counts (2000 HVGs) the model was trained on, from the scverse example data server.Immune_ALL_human.h5ad— the full gene set from the scverse example data server (used only at the end to validate with the held-out marker).
io_dir = Path("./tmp_io/drvi_immune_128/")
embed_path = io_dir / "embed.h5ad"
adata_path = io_dir.parent / "Immune_HVG_human.h5ad"
model_path = io_dir / "drvi_model"
full_anndata_path = io_dir.parent / "Immune_ALL_human.h5ad"
# Column in `embed.obs` / full data holding the existing cell-type annotation.
CELL_TYPE_COL = "final_annotation"
# Thresholds defining a rare-cell-type dimension (see the three criteria above).
ACTIVITY_FRACTION = 0.5 # a cell is "active" if |latent| exceeds this fraction of the dimension's peak |latent|
MAX_RARE_FRACTION = 0.01 # a dimension is "rare" if fewer than this fraction of cells are active
MIN_COHESION = 0.5 # min UMAP cohesion (1 = tight blob, ->0 = spread across the UMAP)
MIN_OOD_SCORE = 1.0 # minimum interpretability score for a gene program
HOST_COVERAGE_CUTOFF = 0.30 # a candidate "needs refinement" if it captures less than this share of its host label
Load artifacts#
embed = sc.read_h5ad(embed_path)
adata = sc.read(
adata_path,
backup_url="https://exampledata.scverse.org/scvi-tools/Immune_HVG_human.h5ad",
)
embed
AnnData object with n_obs × n_vars = 32484 × 128
obs: 'batch', 'chemistry', 'data_type', 'dpt_pseudotime', 'final_annotation', 'mt_frac', 'n_counts', 'n_genes', 'sample_ID', 'size_factors', 'species', 'study', 'tissue', '_scvi_batch', '_scvi_labels'
var: 'original_dim_id', 'reconstruction_effect', 'order', 'max_value', 'mean', 'min', 'max', 'std', 'std_abs', 'title', 'vanished', 'vanished_positive_direction', 'vanished_negative_direction'
uns: 'neighbors', 'pca', 'umap'
obsm: 'X_pca', 'X_umap'
varm: 'IND_exp_weighted_mean_negative', 'IND_exp_weighted_mean_positive', 'IND_linear_weighted_mean_negative', 'IND_linear_weighted_mean_positive', 'IND_max_negative', 'IND_max_positive', 'OOD_combined_negative', 'OOD_combined_positive', 'OOD_max_possible_negative', 'OOD_max_possible_positive', 'OOD_min_possible_negative', 'OOD_min_possible_positive', 'PCs'
obsp: 'connectivities', 'distances'
# Set up and load the trained model. We only need it for the convenience interpretability API
# (get/plot_interpretability_scores); all three criteria are computable from `embed` alone.
DRVI.setup_anndata(
adata,
layer="counts",
batch_key="batch",
is_count_data=True,
)
model = DRVI.load(model_path, adata)
model
INFO File tmp_io/drvi_immune_128/drvi_model/model.pt already downloaded
INFO DRVI: The model is trained with DRVI version 0.2.6.
INFO DRVI: Updaging data setup config ...
INFO DRVI: Done updating data source registry. Loading in DRVI version 0.2.6.
INFO DRVI: Loading model from DRVI version 0.2.6.
INFO DRVI: Done updating model args. Loading in 0.2.6.
INFO DRVI: The model has been initialized
DRVI Latent size: 128, splits: 128, pooling of splits: 'logsumexp', Encoder dims: [128, 128], Decoder dims: [128, 128], Gene likelihood: pnb, Training status: Trained
Scan every dimension for the three criteria#
A few practical points:
DRVI dimension titles do not match the column order in
embed(e.g.DR 1is not the first column), so we always index dimensions by their title.We read interpretability through the model interface,
model.get_interpretability_scores(embed, adata). It returns a genes × dimensions table whose columns are the non-vanished signed dimensions (e.g.DR 1+).
# Set title as index for easier access to latent dimensions by name
embed.var.set_index("title", inplace=True, drop=False)
Utils for criterion 1: number of active cells in a dimension#
def active_mask(embed, dim_title_direction):
"""Boolean mask of cells active on a given dimension/direction.
The threshold is adaptive per dimension: a cell is active when its latent value passes half of
the dimension's peak activation (`max(|latent|) * ACTIVITY_FRACTION`) in the requested direction.
"""
dim_title = dim_title_direction[:-1] # e.g. "DR 49+" -> "DR 49"
direction = dim_title_direction[-1] # e.g. "DR 49+" -> "+"
column = np.asanyarray(embed[:, dim_title].X).flatten()
cutoff = np.abs(column).max() * ACTIVITY_FRACTION # per-dimension: max(|latent|) / 2
return column > cutoff if direction == "+" else column < -cutoff
Utils for criterion 2: spatial cohesion (UMAP compactness vs Leiden clusters)#
A real rare cell type forms a single tight blob rather than being scattered. We measure this directly on the 2-D UMAP — the space we actually visualize. We cluster the latent space with Leiden and take the typical within-cluster pairwise UMAP distance as a reference scale, then compare it to the spread of each dimension’s active cells on the UMAP:
cohesion = reference_cluster_scale / (reference_cluster_scale + median_pairwise_umap_distance_of_active_cells)
This is ~1 for a tight blob and drops toward 0 as the active cells spread out.
sc.tl.leiden(embed, resolution=1.0, key_added="leiden", flavor="igraph", n_iterations=2, directed=False)
leiden_labels = embed.obs["leiden"].to_numpy()
umap = embed.obsm["X_umap"]
def median_pairwise_distance(coords, idx, rng, max_cells=100):
"""Median pairwise distance among `idx` cells in `coords`, sub-sampled to <= max_cells for speed."""
if len(idx) <= 1:
return 0.0
sample = idx if len(idx) <= max_cells else idx[rng.choice(len(idx), max_cells, replace=False)]
return float(np.median(pdist(coords[sample])))
def umap_cohesion(active_idx, coords, cluster_scale, rng):
"""cluster_scale / (cluster_scale + median pairwise UMAP distance); 1 = tight blob, ->0 = spread."""
return cluster_scale / (cluster_scale + median_pairwise_distance(coords, active_idx, rng))
# Reference scale: typical within-Leiden-cluster pairwise UMAP distance.
rng = np.random.RandomState(0)
UMAP_CLUSTER_SCALE = float(np.median([
median_pairwise_distance(umap, np.where(leiden_labels == c)[0], rng)
for c in pd.unique(leiden_labels) if (leiden_labels == c).sum() >= 10
]))
print(f"Leiden: {len(set(leiden_labels))} clusters; "
f"typical within-cluster UMAP spread = {UMAP_CLUSTER_SCALE:.2f}.")
Leiden: 33 clusters; typical within-cluster UMAP spread = 1.35.
Utils for criterion 3: interpretability#
# Interpretability via the model interface: genes x non-vanished signed dimensions (e.g. "DR 49+").
interpretability_df = model.get_interpretability_scores(embed, adata)
interpretability_df.iloc[:3, :4]
| title | DR 1+ | DR 1- | DR 2+ | DR 2- |
|---|---|---|---|---|
| index | ||||
| TCL1A | 0.0 | 0.009215 | 5.813187e-11 | 0.000651 |
| IGLL5 | 0.0 | 0.001222 | 0.000000e+00 | 0.000468 |
| PTGDS | 0.0 | 0.001911 | 1.868569e-08 | 0.000612 |
Calculating all three criteria#
dim_stats = []
for title in embed.var["title"]:
for direction in ["+", "-"]:
dim_title_direction = title + direction
# Find fraction of cells active on this dimension/direction (|latent| beyond the per-dimension cutoff).
active_cells = active_mask(embed, dim_title_direction)
n_active = active_cells.sum()
fraction_active = n_active / embed.n_obs
if n_active == 0:
continue
# find maximum interpretability score for this dimension/direction (i.e. strongest gene program)
if dim_title_direction not in interpretability_df.columns: # Vanished
continue
max_ood_score = interpretability_df[dim_title_direction].max().clip(1e-2, 10) # Clipping for better visualization
# Find UMAP cohesion of active cells (1 = tight blob, ->0 = spread across the UMAP)
rng = np.random.RandomState(0)
cohesion = umap_cohesion(np.where(active_cells)[0], umap, UMAP_CLUSTER_SCALE, rng)
dim_stats.append(
{
"dim_title_direction": dim_title_direction,
"n_active": n_active,
"fraction_active": fraction_active,
"max_ood_score": max_ood_score,
"cohesion": cohesion,
}
)
dim_stats = pd.DataFrame(dim_stats).set_index("dim_title_direction")
dim_stats.sort_values("fraction_active")[:10]
| n_active | fraction_active | max_ood_score | cohesion | |
|---|---|---|---|---|
| dim_title_direction | ||||
| DR 6- | 1 | 0.000031 | 0.010000 | 1.000000 |
| DR 39- | 2 | 0.000062 | 0.010000 | 0.082661 |
| DR 56+ | 6 | 0.000185 | 5.506810 | 0.106855 |
| DR 49+ | 11 | 0.000339 | 10.000000 | 0.950921 |
| DR 53+ | 17 | 0.000523 | 7.863611 | 0.893626 |
| DR 52- | 19 | 0.000585 | 1.186558 | 0.917455 |
| DR 47- | 31 | 0.000954 | 9.252443 | 0.903657 |
| DR 51- | 39 | 0.001201 | 6.611657 | 0.682976 |
| DR 50- | 46 | 0.001416 | 0.362388 | 0.806846 |
| DR 55- | 47 | 0.001447 | 1.327584 | 0.527321 |
Selecting candidates#
We now have all three per-dimension quantities: rarity (fraction, criterion 1), spatial cohesion (criterion 2), and interpretability (criterion 3, the max OOD score). A dimension is a rare-cell-type candidate when it passes all three thresholds.
dim_stats["is_candidate"] = (
(dim_stats["fraction_active"] < MAX_RARE_FRACTION)
& (dim_stats["cohesion"] > MIN_COHESION)
& (dim_stats["max_ood_score"] > MIN_OOD_SCORE)
)
print(f"{dim_stats['is_candidate'].sum()} candidates satisfy all three criteria.")
21 candidates satisfy all three criteria.
Ranking dimensions by a combined score#
We combine the three criteria into a single score for every dimension: each is normalized to [0, 1] and
the three are multiplied, so a dimension must be rare and compact and interpretable to rank high.
def norm01(x):
return (x - x.min()) / (x.max() - x.min())
dim_stats["rarity_score"] = norm01(-np.log10(dim_stats["fraction_active"])) # rarer -> higher
dim_stats["cohesion_score"] = dim_stats["cohesion"] # already 0-1
dim_stats["interpretability_score"] = norm01(np.log10(dim_stats["max_ood_score"])) # stronger -> higher
dim_stats["score"] = (
dim_stats["rarity_score"] * dim_stats["cohesion_score"] * dim_stats["interpretability_score"]
)
dim_stats = dim_stats.sort_values("score", ascending=False)
dim_stats[["n_active", "fraction_active", "cohesion", "max_ood_score", "score", "is_candidate"]]
| n_active | fraction_active | cohesion | max_ood_score | score | is_candidate | |
|---|---|---|---|---|---|---|
| dim_title_direction | ||||||
| DR 49+ | 11 | 0.000339 | 0.950921 | 10.000000 | 0.685543 | True |
| DR 53+ | 17 | 0.000523 | 0.893626 | 7.863611 | 0.578123 | True |
| DR 47- | 31 | 0.000954 | 0.903657 | 9.252443 | 0.536401 | True |
| DR 52- | 19 | 0.000585 | 0.917455 | 1.186558 | 0.416973 | True |
| DR 51- | 39 | 0.001201 | 0.682976 | 6.611657 | 0.368305 | True |
| DR 26+ | 139 | 0.004279 | 0.767048 | 9.403327 | 0.323632 | True |
| DR 42+ | 106 | 0.003263 | 0.682977 | 10.000000 | 0.312294 | True |
| DR 41+ | 130 | 0.004002 | 0.773856 | 5.237635 | 0.304060 | True |
| DR 25+ | 189 | 0.005818 | 0.695459 | 5.538980 | 0.248000 | True |
| DR 16- | 287 | 0.008835 | 0.733257 | 8.359651 | 0.243792 | True |
| DR 46+ | 133 | 0.004094 | 0.720604 | 2.203845 | 0.242495 | True |
| DR 38+ | 104 | 0.003202 | 0.683120 | 1.912337 | 0.238708 | True |
| DR 50- | 46 | 0.001416 | 0.806846 | 0.362388 | 0.232485 | False |
| DR 55- | 47 | 0.001447 | 0.527321 | 1.327584 | 0.205960 | True |
| DR 31+ | 150 | 0.004618 | 0.587418 | 3.207656 | 0.204557 | True |
| DR 36- | 168 | 0.005172 | 0.603192 | 3.123841 | 0.202471 | True |
| DR 21- | 278 | 0.008558 | 0.623853 | 5.030608 | 0.193845 | True |
| DR 30- | 199 | 0.006126 | 0.512849 | 4.541783 | 0.174409 | True |
| DR 35+ | 213 | 0.006557 | 0.557321 | 1.598565 | 0.153947 | True |
| DR 43- | 96 | 0.002955 | 0.317666 | 10.000000 | 0.148917 | False |
| DR 20- | 293 | 0.009020 | 0.547869 | 2.481305 | 0.148219 | True |
| DR 34+ | 248 | 0.007635 | 0.531915 | 1.605116 | 0.140124 | True |
| DR 23+ | 456 | 0.014038 | 0.593526 | 2.542973 | 0.136788 | False |
| DR 40- | 207 | 0.006372 | 0.509244 | 1.235535 | 0.134706 | True |
| DR 19- | 398 | 0.012252 | 0.562408 | 1.695194 | 0.126743 | False |
| DR 28- | 403 | 0.012406 | 0.504199 | 2.583121 | 0.122360 | False |
| DR 37+ | 69 | 0.002124 | 0.417962 | 0.512963 | 0.120845 | False |
| DR 17+ | 531 | 0.016347 | 0.590460 | 1.847625 | 0.120327 | False |
| DR 22- | 477 | 0.014684 | 0.537213 | 2.381028 | 0.120109 | False |
| DR 24- | 428 | 0.013176 | 0.561466 | 1.432313 | 0.118964 | False |
| DR 10+ | 822 | 0.025305 | 0.521597 | 10.000000 | 0.114160 | False |
| DR 32- | 386 | 0.011883 | 0.434243 | 3.509716 | 0.113047 | False |
| DR 27- | 465 | 0.014315 | 0.534987 | 1.563585 | 0.111580 | False |
| DR 44+ | 101 | 0.003109 | 0.469374 | 0.343807 | 0.111261 | False |
| DR 14+ | 507 | 0.015608 | 0.493000 | 1.740805 | 0.101302 | False |
| DR 15- | 524 | 0.016131 | 0.503970 | 1.176124 | 0.094351 | False |
| DR 7- | 1113 | 0.034263 | 0.501954 | 5.309289 | 0.083709 | False |
| DR 9- | 1451 | 0.044668 | 0.547161 | 10.000000 | 0.083567 | False |
| DR 29+ | 459 | 0.014130 | 0.398992 | 1.234260 | 0.079741 | False |
| DR 56+ | 6 | 0.000185 | 0.106855 | 5.506810 | 0.077268 | False |
| DR 13+ | 966 | 0.029738 | 0.515752 | 1.432883 | 0.074167 | False |
| DR 8- | 1202 | 0.037003 | 0.470058 | 3.136298 | 0.068311 | False |
| DR 18- | 988 | 0.030415 | 0.466583 | 1.354993 | 0.065472 | False |
| DR 5+ | 1463 | 0.045038 | 0.479768 | 4.922354 | 0.065343 | False |
| DR 3+ | 1606 | 0.049440 | 0.463542 | 10.000000 | 0.065321 | False |
| DR 48- | 303 | 0.009328 | 0.176518 | 3.737558 | 0.050711 | False |
| DR 33- | 93 | 0.002863 | 0.106570 | 10.000000 | 0.050352 | False |
| DR 45+ | 207 | 0.006372 | 0.147329 | 4.121349 | 0.048719 | False |
| DR 12- | 1152 | 0.035464 | 0.381117 | 1.292598 | 0.048171 | False |
| DR 4- | 1418 | 0.043652 | 0.417651 | 1.290344 | 0.045666 | False |
| DR 2- | 1803 | 0.055504 | 0.352871 | 10.000000 | 0.044974 | False |
| DR 11- | 1306 | 0.040204 | 0.387866 | 0.903089 | 0.041716 | False |
| DR 6+ | 2662 | 0.081948 | 0.365708 | 1.969929 | 0.022965 | False |
| DR 39+ | 357 | 0.010990 | 0.125989 | 0.068013 | 0.011047 | False |
| DR 6- | 1 | 0.000031 | 1.000000 | 0.010000 | 0.000000 | False |
| DR 39- | 2 | 0.000062 | 0.082661 | 0.010000 | 0.000000 | False |
| DR 1- | 5390 | 0.165928 | 0.390798 | 7.170271 | 0.000000 | False |
| DR 1+ | 106 | 0.003263 | 0.121064 | 0.010000 | 0.000000 | False |
| DR 54+ | 179 | 0.005510 | 0.110969 | 0.010000 | 0.000000 | False |
Combined score vs each criterion#
The combined score is the product of the three per-criterion scores, so we plot it against each one. Every dimension is shown; candidates (passing all three thresholds) are red, and the top 5 by combined score are labeled. A candidate scores highly on all three axes; a dimension weak on any single criterion is pulled down.
top5 = dim_stats.nlargest(5, "score")
components = [
("rarity_score", "Rarity score"),
("cohesion_score", "Cohesion score"),
("interpretability_score", "Interpretability score"),
]
cand = dim_stats[dim_stats["is_candidate"]]
noncand = dim_stats[~dim_stats["is_candidate"]]
fig, axes = plt.subplots(1, 3, figsize=(12, 3.6), sharey=True)
for ax, (col, label) in zip(axes, components):
ax.scatter(noncand[col], noncand["score"], s=25, c="lightgray")
ax.scatter(cand[col], cand["score"], s=35, c="crimson", label="candidate")
for dim, r in top5.iterrows():
ax.annotate(dim, (r[col], r["score"]), xytext=(4, 4), textcoords="offset points", fontsize=7)
ax.set_xlabel(label)
axes[0].set_ylabel("Combined score")
axes[0].legend(frameon=False, fontsize=8, loc="upper left")
fig.suptitle("Combined score vs each criterion")
plt.tight_layout()
plt.show()
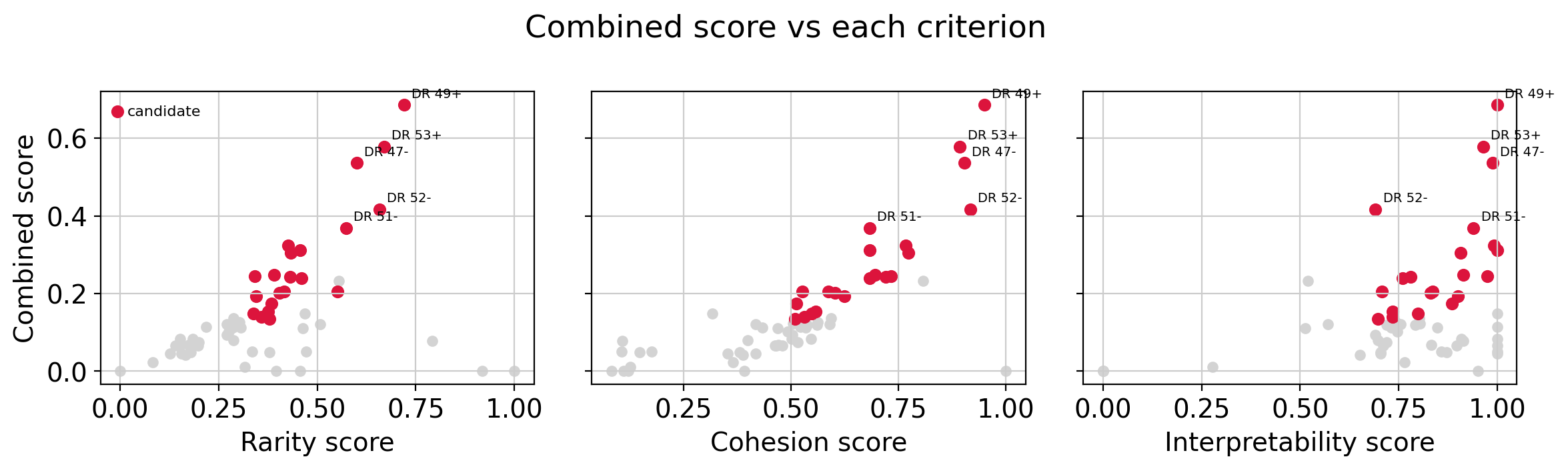
Which candidates are good rare-cell-type candidates?#
Now we bring in the existing annotation (final_annotation) — but the question is not whether a
dimension’s active cells carry one label or several. A rare type whose cells are all labeled X is no
more or less interesting than one labeled half X, half Y; both can be perfectly valid known types.
What signals a hidden population that needs refinement is different: the active cells are labeled X,
yet they make up only a small fraction of all X cells (say < 30%). That means the dimension has
carved out a distinct minority sub-population inside an existing annotation — a cell state the label does
not resolve. So for each candidate we compute its host-annotation coverage: of all cells carrying the
candidate’s dominant label, what fraction are active on this dimension.
High coverage (≈100%) → the dimension simply re-discovers a known annotated type.
Low coverage (
< 30%) → a sub-population hidden inside that annotation — a refinement candidate.
# Now we finally subset to the candidates (all three criteria) and characterize them by annotation.
candidates = dim_stats[dim_stats["is_candidate"]].copy()
annotation = embed.obs[CELL_TYPE_COL].astype(str).values
annotation_totals = pd.Series(annotation).value_counts()
def host_annotation_coverage(embed, dim_title_direction, annotation, annotation_totals):
"""Dominant label of the active cells, and the fraction of *all* cells with that label
that this dimension captures."""
active = active_mask(embed, dim_title_direction)
counts = pd.Series(annotation[active]).value_counts()
host = counts.index[0]
return host, counts.iloc[0] / annotation_totals[host]
coverage = [host_annotation_coverage(embed, dim_title_direction, annotation, annotation_totals)
for dim_title_direction in candidates.index]
candidates["host_annotation"] = [host for host, _ in coverage]
candidates["host_coverage"] = [cov for _, cov in coverage]
Sorting the candidates by host-annotation coverage: dimensions on the right re-discover whole annotated types (e.g. one capturing nearly all Plasma cells, another nearly all pDCs), while those below the 30% cutoff are minority sub-populations hidden inside a label. The low-coverage candidates are the good rare-cell-type candidates — cell states the current annotation does not resolve.
cov_sorted = candidates.sort_values("host_coverage")
colors = ["crimson" if c < HOST_COVERAGE_CUTOFF else "lightgray" for c in cov_sorted["host_coverage"]]
fig, ax = plt.subplots(figsize=(max(5.0, 0.28 * len(cov_sorted)), 4))
ax.bar(cov_sorted.index, cov_sorted["host_coverage"], color=colors)
ax.axhline(HOST_COVERAGE_CUTOFF, ls="--", c="gray", lw=1, label=f"{HOST_COVERAGE_CUTOFF:.0%} cutoff")
ax.set_ylabel("Host-annotation coverage")
ax.set_xlabel("Candidate dimension (sorted)")
ax.set_title("Fraction of the host annotation captured by each candidate")
ax.tick_params(axis="x", rotation=90)
ax.legend(frameon=False, fontsize=8)
plt.tight_layout()
plt.show()
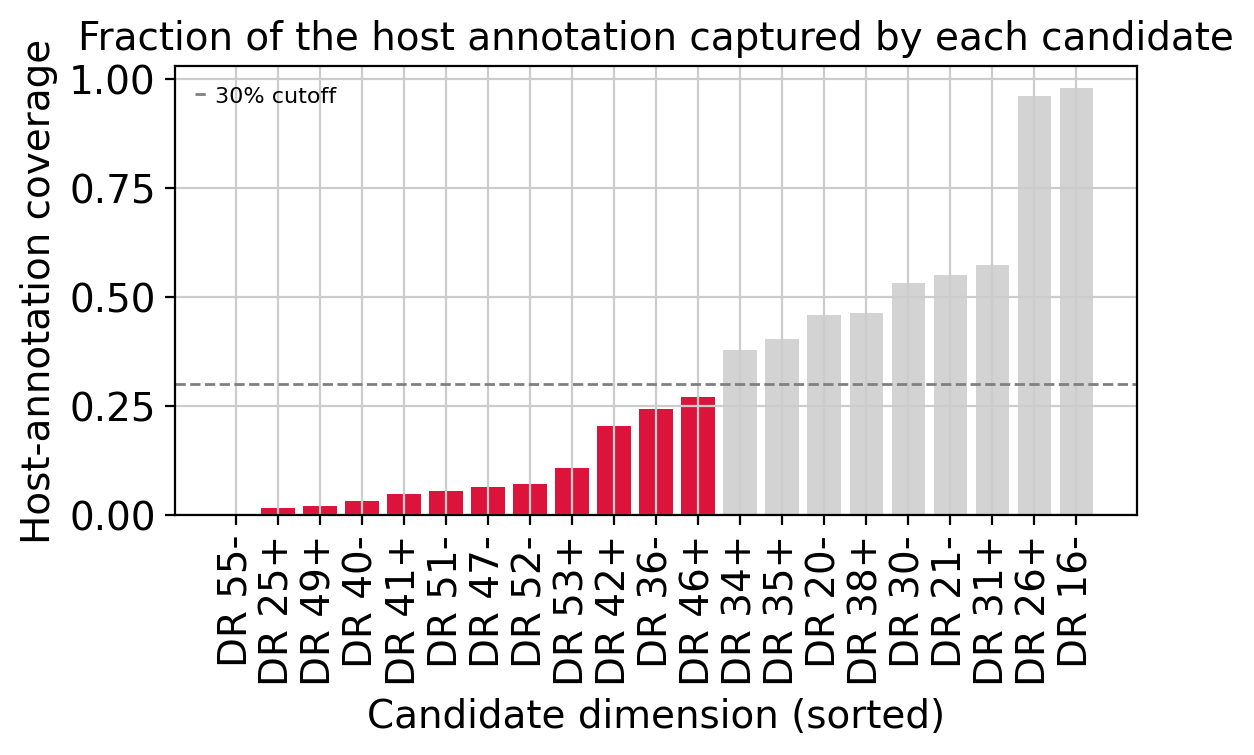
unannotated_candidates = candidates[candidates["host_coverage"] < HOST_COVERAGE_CUTOFF]
print(f"{len(unannotated_candidates)} unannotated candidates (host coverage < {HOST_COVERAGE_CUTOFF:.0%}).")
unannotated_candidates
12 unannotated candidates (host coverage < 30%).
| n_active | fraction_active | max_ood_score | cohesion | is_candidate | rarity_score | cohesion_score | interpretability_score | score | host_annotation | host_coverage | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| dim_title_direction | |||||||||||
| DR 49+ | 11 | 0.000339 | 10.000000 | 0.950921 | True | 0.720925 | 0.950921 | 1.000000 | 0.685543 | HSPCs | 0.021142 |
| DR 53+ | 17 | 0.000523 | 7.863611 | 0.893626 | True | 0.670261 | 0.893626 | 0.965207 | 0.578123 | Plasma cells | 0.108527 |
| DR 47- | 31 | 0.000954 | 9.252443 | 0.903657 | True | 0.600341 | 0.903657 | 0.988752 | 0.536401 | Monocyte-derived dendritic cells | 0.064854 |
| DR 52- | 19 | 0.000585 | 1.186558 | 0.917455 | True | 0.657317 | 0.917455 | 0.691430 | 0.416973 | Megakaryocyte progenitors | 0.070370 |
| DR 51- | 39 | 0.001201 | 6.611657 | 0.682976 | True | 0.573623 | 0.682976 | 0.940103 | 0.368305 | Monocyte-derived dendritic cells | 0.054393 |
| DR 42+ | 106 | 0.003263 | 10.000000 | 0.682977 | True | 0.457254 | 0.682977 | 1.000000 | 0.312294 | Monocyte progenitors | 0.203271 |
| DR 41+ | 130 | 0.004002 | 5.237635 | 0.773856 | True | 0.433500 | 0.773856 | 0.906378 | 0.304060 | NK cells | 0.047515 |
| DR 25+ | 189 | 0.005818 | 5.538980 | 0.695459 | True | 0.389948 | 0.695459 | 0.914477 | 0.248000 | CD4+ T cells | 0.016620 |
| DR 46+ | 133 | 0.004094 | 2.203845 | 0.720604 | True | 0.430845 | 0.720604 | 0.781060 | 0.242495 | Monocyte-derived dendritic cells | 0.269874 |
| DR 55- | 47 | 0.001447 | 1.327584 | 0.527321 | True | 0.551907 | 0.527321 | 0.707687 | 0.205960 | CD4+ T cells | 0.002997 |
| DR 36- | 168 | 0.005172 | 3.123841 | 0.603192 | True | 0.403656 | 0.603192 | 0.831563 | 0.202471 | Monocyte progenitors | 0.242991 |
| DR 40- | 207 | 0.006372 | 1.235535 | 0.509244 | True | 0.379361 | 0.509244 | 0.697285 | 0.134706 | CD14+ Monocytes | 0.031398 |
Inspect the good candidates#
For every good rare-cell-type candidate we look at where its active cells sit in the latent UMAP and which genes define it. Each candidate should light up a small, coherent region (cohesion) with a distinct gene program (interpretability).
candidate_dims = unannotated_candidates.index.to_list()
drvi.utils.pl.plot_latent_dims_in_umap(embed, dim_subset=candidate_dims, directional=True, ncols=5)
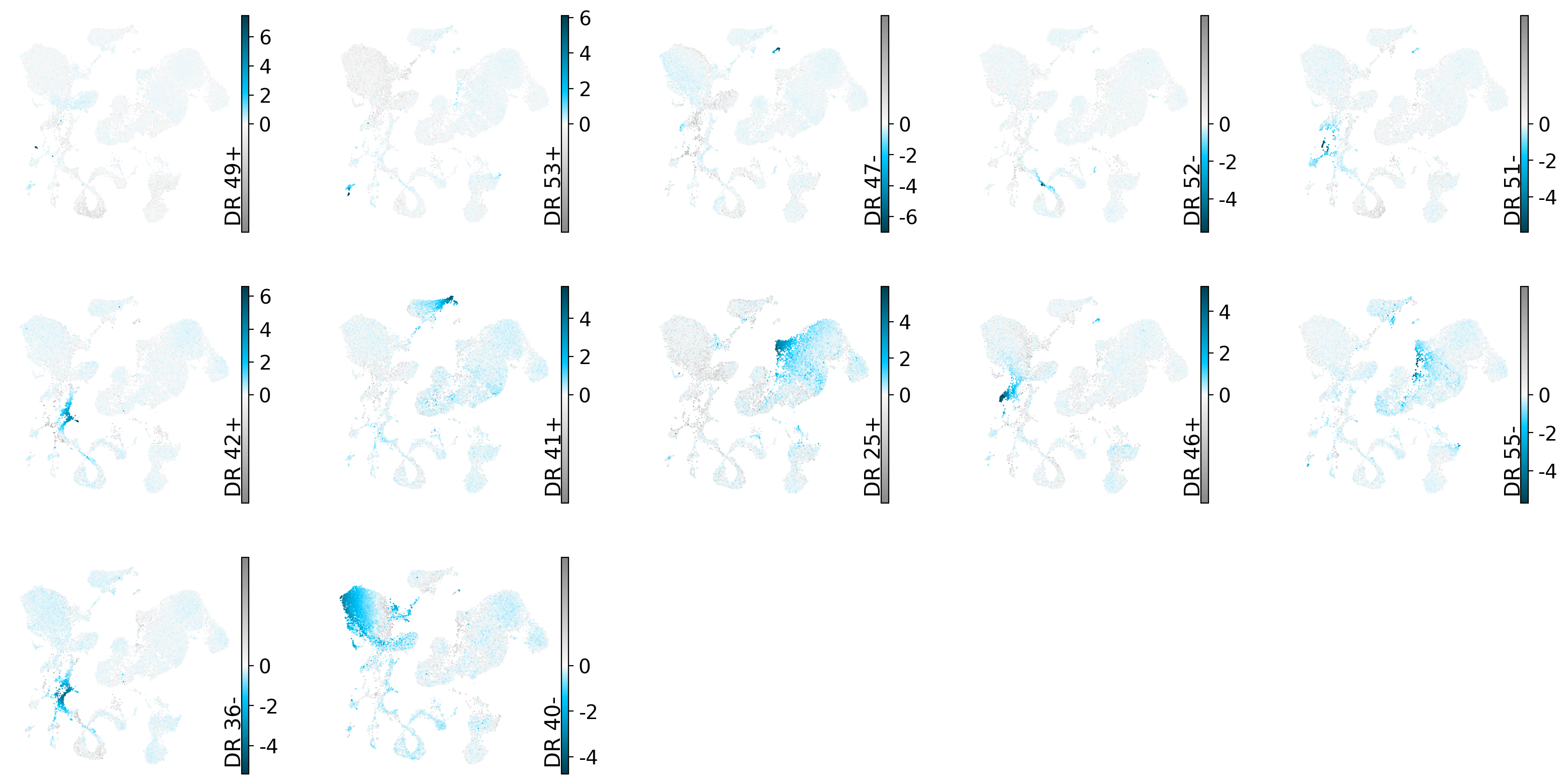
# `interpretability_df` (from get_interpretability_scores) already holds the gene x dimension scores;
# we plot it directly with the utility function.
drvi.utils.pl.plot_interpretability_scores(interpretability_df, dim_subset=candidate_dims)
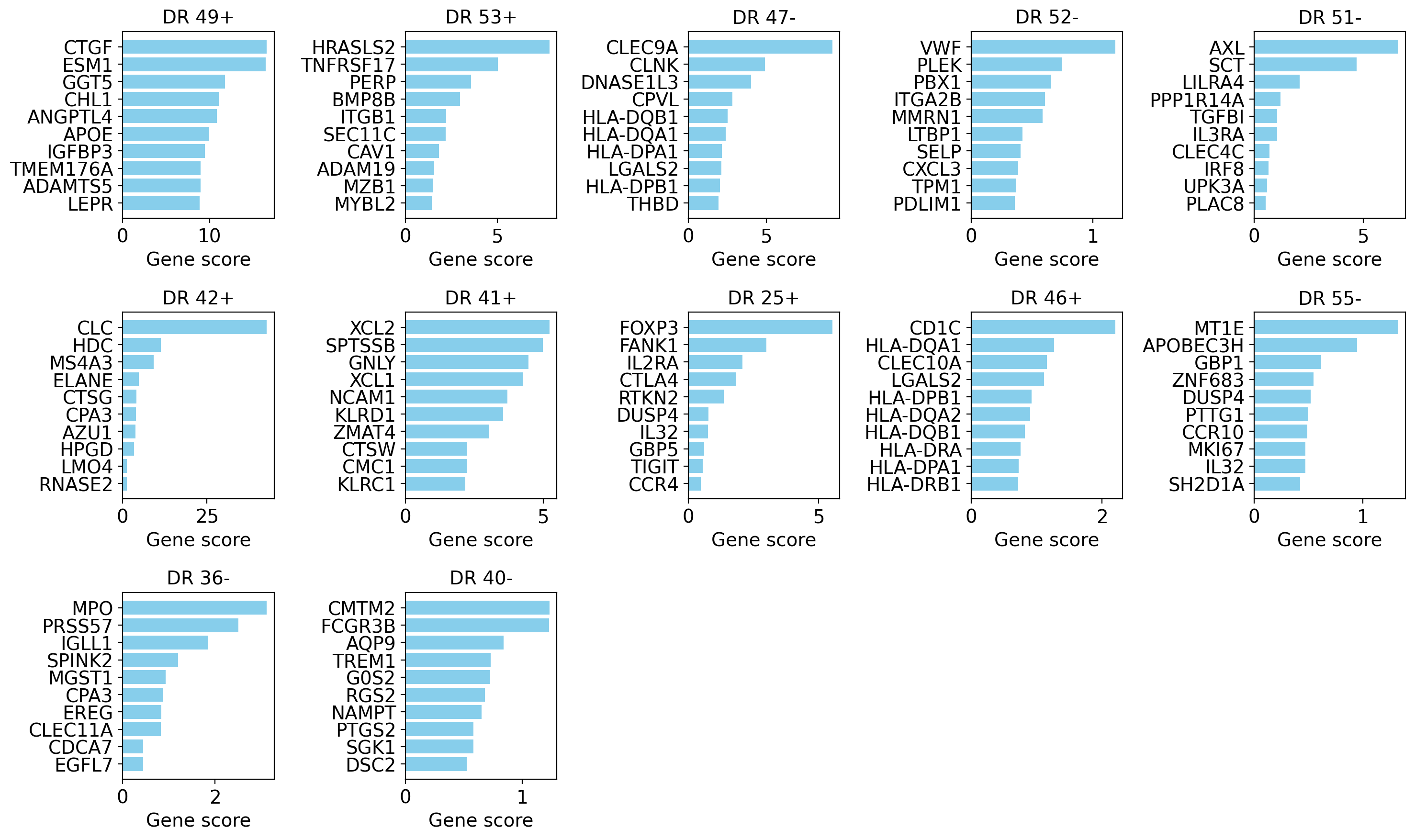
The candidates above represent rare cell types or substantially finer subclusters than the existing annotations. These subsets can usually be identified from their top marker genes.
A closer look: discovering the fibroblasts#
Among the candidates, the rarest one stands out: a tiny dimension (about a dozen cells) whose cells are
filed under HSPCs yet are only ~2% of all HSPCs. Let us follow it end-to-end — identify it, read its gene
program, and prove what it is.
novel_dim = unannotated_candidates["fraction_active"].idxmin() # signed title of the rarest unannotated candidate
novel = unannotated_candidates.loc[novel_dim]
print(f"Rarest candidate: {novel_dim} "
f"({int(novel['n_active'])} active cells, "
f"filed under '{novel['host_annotation']}' = {novel['host_coverage']:.0%} of that label)")
Rarest candidate: DR 49+ (11 active cells, filed under 'HSPCs' = 2% of that label)
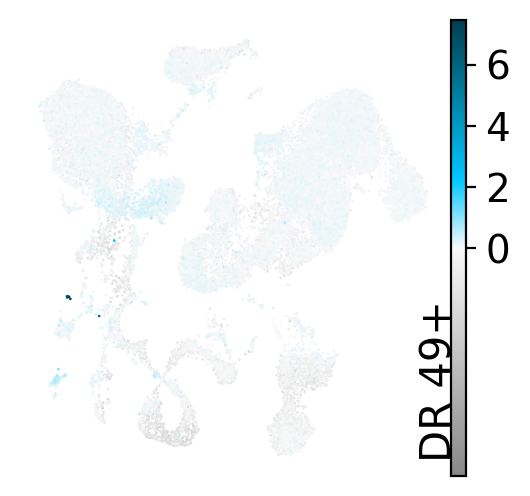
Where do these cells sit in the latent UMAP, and what gene program defines the dimension?
drvi.utils.pl.plot_latent_dims_in_umap(embed, dim_subset=[novel_dim], directional=True)
# Top genes of the dimension from the OOD interpretability scores computed earlier.
top_genes = interpretability_df[novel_dim].sort_values(ascending=False).head(15)
top_genes
index
CTGF 16.571638
ESM1 16.453249
GGT5 11.744926
CHL1 11.075065
ANGPTL4 10.839224
APOE 9.956368
IGFBP3 9.464329
TMEM176A 8.986178
ADAMTS5 8.935861
LEPR 8.833095
PCOLCE 8.585528
TNFAIP6 8.509926
APOD 8.253371
CYP1B1 8.215649
SOCS3 7.993901
Name: DR 49+, dtype: float32
drvi.utils.pl.plot_interpretability_scores(interpretability_df, dim_subset=[novel_dim])
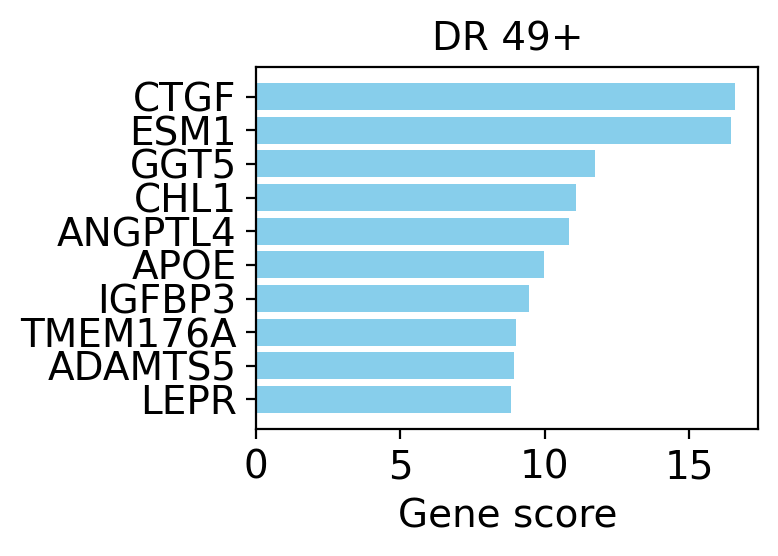
The program is dominated by extracellular-matrix / stromal genes — CTGF (CCN2), PCOLCE, ADAMTS5, IGFBP3, APOD, ANGPTL4 — none of which belong to immune cells. The hypothesis is clear: these few cells are fibroblasts, a stromal contaminant that the annotation merged into HSPCs.
Crucially, the canonical fibroblast marker COL1A1 is not in the 2000 HVGs, so the model never saw
it. That makes it a perfect independent test of the hypothesis.
Validate with the held-out marker COL1A1#
We load the full gene set, label the active cells of the novel dimension as Fibroblast (DRVI), and show
that COL1A1 lights up specifically in this group — and not in the HSPCs they were mislabeled as.
# The full matrix (~2 GB) is only needed here.
full = sc.read(
full_anndata_path,
backup_url="https://exampledata.scverse.org/scvi-tools/Immune_ALL_human.h5ad",
)
full = full[embed.obs_names].copy()
assert "COL1A1" not in adata.var_names, "COL1A1 should be held out from the HVGs the model trained on"
assert "COL1A1" in full.var_names, "COL1A1 must be present in the full gene set"
# Ensure a log-normalized layer for the dotplot.
full.layers["log1p"] = full.layers["counts"].copy()
full.X = full.layers["log1p"]
sc.pp.normalize_total(full)
sc.pp.log1p(full)
full.layers["log1p"] = full.X.copy()
# Label the active cells of the novel dimension; everyone else keeps their original annotation.
active_novel = active_mask(embed, novel_dim)
full.obs["new_cell_type"] = np.where(
active_novel, "Fibroblast (DRVI)", full.obs[CELL_TYPE_COL].astype(str)
)
print(full.obs["new_cell_type"].value_counts().loc[["Fibroblast (DRVI)"]])
new_cell_type
Fibroblast (DRVI) 11
Name: count, dtype: int64
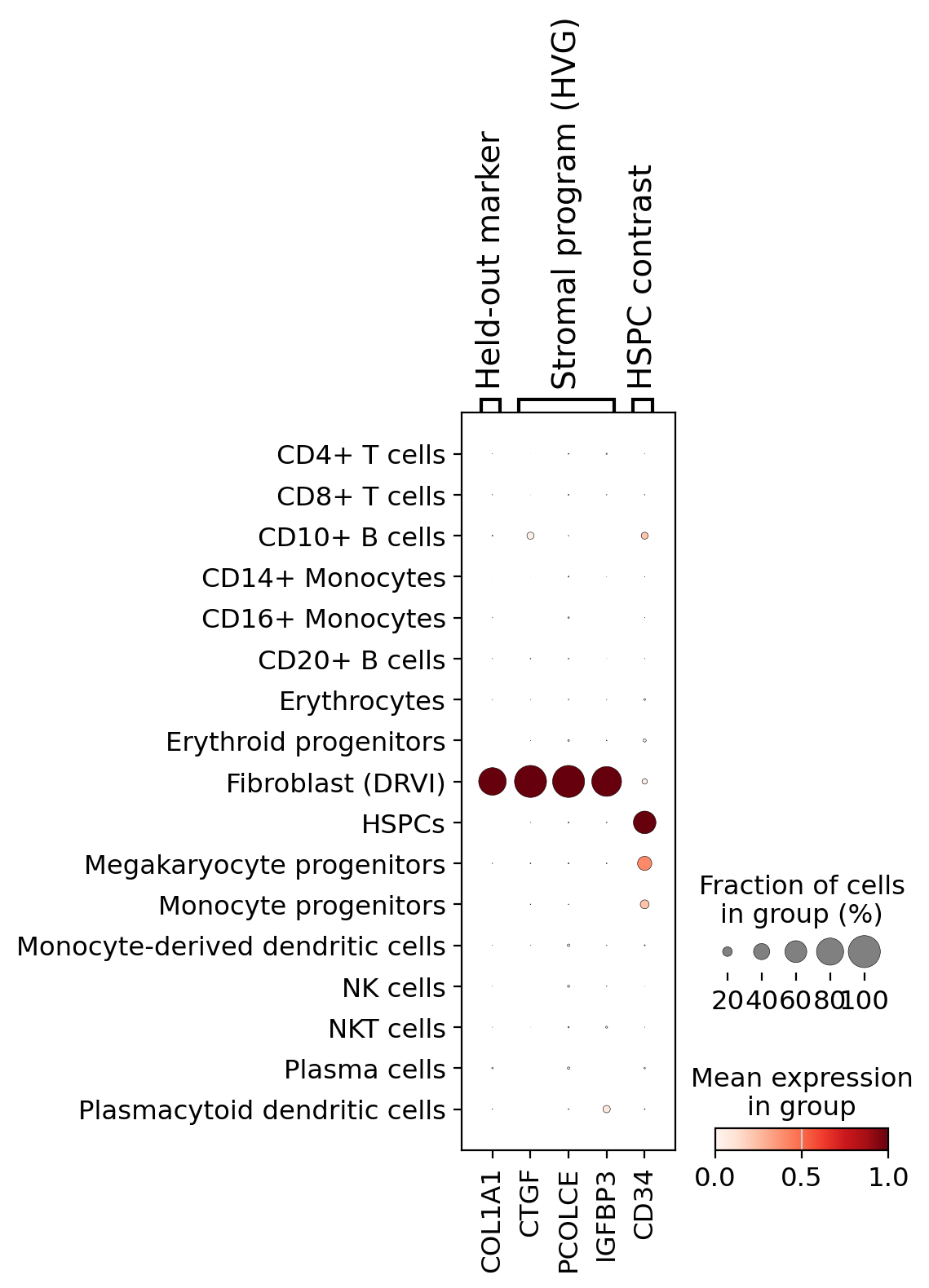
Marker dotplot. COL1A1 is the held-out proof; the stromal HVG genes are the program DRVI used; CD34
is shown as a contrast marker for the HSPCs these cells were labeled as.
marker_genes = {
"Held-out marker": ["COL1A1"],
"Stromal program (HVG)": ["CTGF", "PCOLCE", "IGFBP3"],
"HSPC contrast": ["CD34"],
}
sc.pl.dotplot(
full,
marker_genes,
groupby="new_cell_type",
standard_scale="var",
layer="log1p",
show=True,
)
Quantitative proof#
col1a1 = np.asarray(
full[:, "COL1A1"].layers["counts"].todense()
if hasattr(full[:, "COL1A1"].layers["counts"], "todense")
else full[:, "COL1A1"].layers["counts"]
).ravel()
frac_in_dim = (col1a1[active_novel] > 0).mean()
n_total_pos = int((col1a1 > 0).sum())
print(f"COL1A1+ among the {int(active_novel.sum())} cells of {novel_dim}: {frac_in_dim:.0%}")
print(f"COL1A1+ cells in the whole dataset: {n_total_pos} / {full.n_obs}")
COL1A1+ among the 11 cells of DR 49+: 82%
COL1A1+ cells in the whole dataset: 67 / 32484
A large majority of the dimension’s cells express COL1A1, while only ~70 cells express it across the
entire dataset (of >32,000) — overwhelming, independent confirmation that DRVI isolated the fibroblasts.
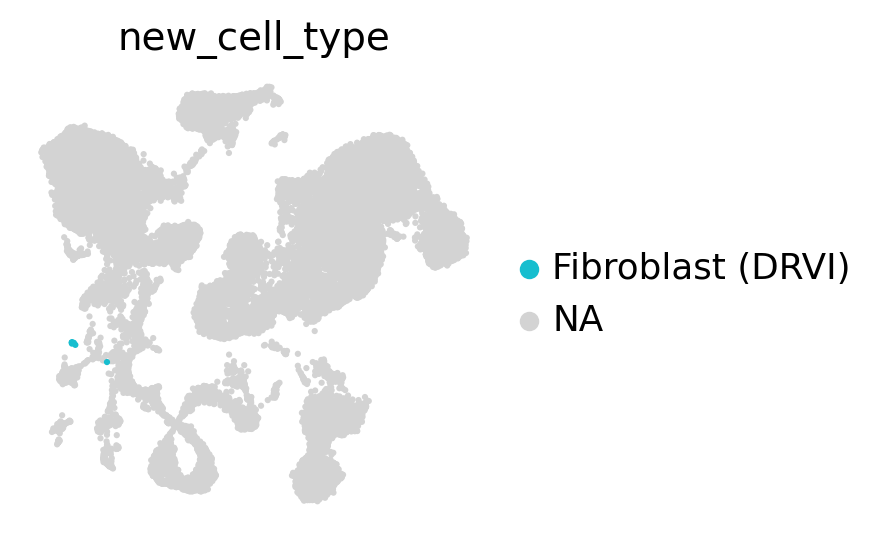
Where are the fibroblasts in the UMAP?#
embed.obs["new_cell_type"] = full.obs["new_cell_type"].values
sc.pl.umap(embed, color="new_cell_type", groups=["Fibroblast (DRVI)"], size=20, frameon=False)
Wrap-up#
Rare, un-annotated populations surface in DRVI as dedicated latent dimensions. The recipe:
Rarity & interpretability — few active cells (
|latent|past half the dimension’s peak) and a specific gene program (max OOD score> 1).Spatial cohesion — the active cells form a tight blob on the UMAP (high UMAP cohesion).
Spot the ones needing refinement — candidates that capture only a small fraction (
< 30%) of their host annotation are hidden sub-populations the labels do not resolve.Propose an identity — read the dimension’s top interpretability genes.
Confirm it [optional] — with an independent, held-out marker (
COL1A1for fibroblasts here).
All thresholds (the activity cutoff, the rare-fraction, cohesion, OOD, and host-coverage cutoffs) are
tunable. Here the activity cutoff is adaptive — half of each dimension’s peak |latent|
(max(|value|) / 2), which self-calibrates per dimension; a fixed cutoff such as ±2 is a common
alternative. The same recipe works for any rare population in any DRVI model — the immune fibroblasts are
just a clean illustration.